From Brain Signal to Muscle Contraction: The Full Mechanism
Every time you move, whether it’s a maximal sprint or reaching for a glass of water, your brain sends a…

By
March 5, 2026
Every time you move, whether it’s a maximal sprint or reaching for a glass of water, your brain sends a signal that travels through your nervous system, crosses a gap between a nerve and a muscle, triggers a cascade of ion movements, releases calcium, and ultimately causes microscopic protein filaments to slide past each other. All of that happens in milliseconds.
Understanding this process in detail isn’t just interesting. It’s foundational to understanding fatigue, training adaptation, and how the body manages energy during exercise. So let’s walk through it from the beginning.
The Resting Membrane Potential: Building the Charge Difference
Before a signal can travel anywhere, the cell has to be set up to carry one. That setup is the resting membrane potential, a voltage difference across the cell membrane, with the inside of the cell negative relative to the outside.
This gradient exists because of differences in ion concentrations on either side of the membrane, primarily potassium (K⁺) and sodium (Na⁺). Those concentration differences are created and maintained by the sodium-potassium ATPase pump, which continuously pumps three sodium ions out of the cell for every two potassium ions it brings in, using ATP to do so.
The result is a steep concentration gradient: potassium is concentrated inside the cell at around 150 millimolar, while outside it’s only about 5 millimolar. Sodium is the reverse, about 145 millimolar outside and 15 millimolar inside.
Inside the cell, large negatively charged molecules, including proteins, ATP (which carries a 4− charge), ADP (3−), and others, cannot cross the membrane. They stay put and keep the interior electrically negative.
Now, potassium follows its concentration gradient and wants to leave the cell through potassium leak channels. But as it leaves, it creates an even more negative interior, which starts pulling it back in. The voltage at which those two forces, the concentration gradient pushing it out and the electrical gradient pulling it back, reach balance can be calculated using the Nernst equation:
E = (61/Z) × log([ion]outside / [ion]inside)
For potassium, plugging in 5 millimolar outside and 150 millimolar inside gives approximately −90 millivolts. For sodium, the same calculation gives about +60 millivolts.
In a neuron, a small amount of sodium leaks in through sodium leak channels, causing a slight depolarization. The result is a resting membrane potential of approximately −70 millivolts, not quite the −90 millivolt potassium equilibrium, because sodium is partially counteracting it. Skeletal muscle cells sit closer to −90 millivolts.
A more complete calculation that accounts for all ions and their relative permeabilities uses the Goldman-Hodgkin-Katz equation, which arrives at exactly the measured values for specific cell types.
Generating an Action Potential: EPSPs, IPSPs, and Summation
With the resting potential established, the neuron is primed and ready. But firing requires reaching a threshold.
Incoming signals arrive at the dendrites and soma of the neuron in two forms. Excitatory postsynaptic potentials (EPSPs) depolarize the membrane. For example, the neurotransmitter glutamate binds to ligand-gated ion channels and allows sodium to flow in, pushing the membrane potential in a positive direction. Inhibitory postsynaptic potentials (IPSPs) do the opposite. Neurotransmitters like GABA or glycine open chloride channels (or potassium channels), driving the membrane potential more negative.
All of these inputs are integrated at the axon hillock. If the net result reaches the threshold of approximately −55 millivolts, an action potential fires. If not, the signal dissipates.
Two forms of summation can help reach that threshold. Temporal summation occurs when the same neuron fires repeatedly in quick succession, with each signal adding to the last before it fades. Spatial summation occurs when multiple neurons fire simultaneously, combining their individual contributions to push the membrane potential over threshold.
The Action Potential Itself: Voltage-Gated Channels in Sequence
Once −55 millivolts is reached, the voltage-gated sodium channels at the axon hillock respond. These channels have two gates, an M gate (activation gate) and an H gate (inactivation gate).
At −55 millivolts, the M gate opens rapidly and sodium rushes into the cell, causing fast depolarization up to about +30 millivolts. At that point, the H gate closes, blocking further sodium entry. Meanwhile, triggered by the same −55 millivolt threshold but more slowly, voltage-gated potassium channels open. Potassium rushes out, rapidly repolarizing the membrane.
The potassium channels are slow to close, so the membrane continues past −70 millivolts down to about −90 millivolts before the system resets, producing a brief hyperpolarization. Sodium leak channels then gradually bring the potential back to −70 millivolts.
This timing creates two distinct refractory periods. During the absolute refractory period, from depolarization through repolarization until the H gates begin reopening at −70 millivolts, no stimulus of any strength can fire another action potential, because the sodium channel inactivation gates are still closed. During the relative refractory period, from −70 back toward resting potential, a sufficiently strong stimulus can trigger another action potential, but it requires more than usual because the cell is hyperpolarized and more of the sodium channels are still recovering.
Saltatory Conduction: Speeding the Signal Down the Axon
The action potential now needs to travel down the axon to reach the muscle. In myelinated neurons, Schwann cells (in the peripheral nervous system) wrap segments of the axon in myelin, an insulating layer that dramatically increases electrical resistance across those segments. Sodium channels are clustered only at the gaps between myelin segments, called nodes of Ranvier.
Rather than traveling continuously along the membrane, the depolarization jumps from node to node in a process called saltatory conduction (from the Latin saltare, to jump). This makes transmission hundreds of times faster and more energy-efficient than in unmyelinated fibers. The axon itself is also narrow and stripped of organelles, allowing the electrical signal to travel with minimal charge loss.
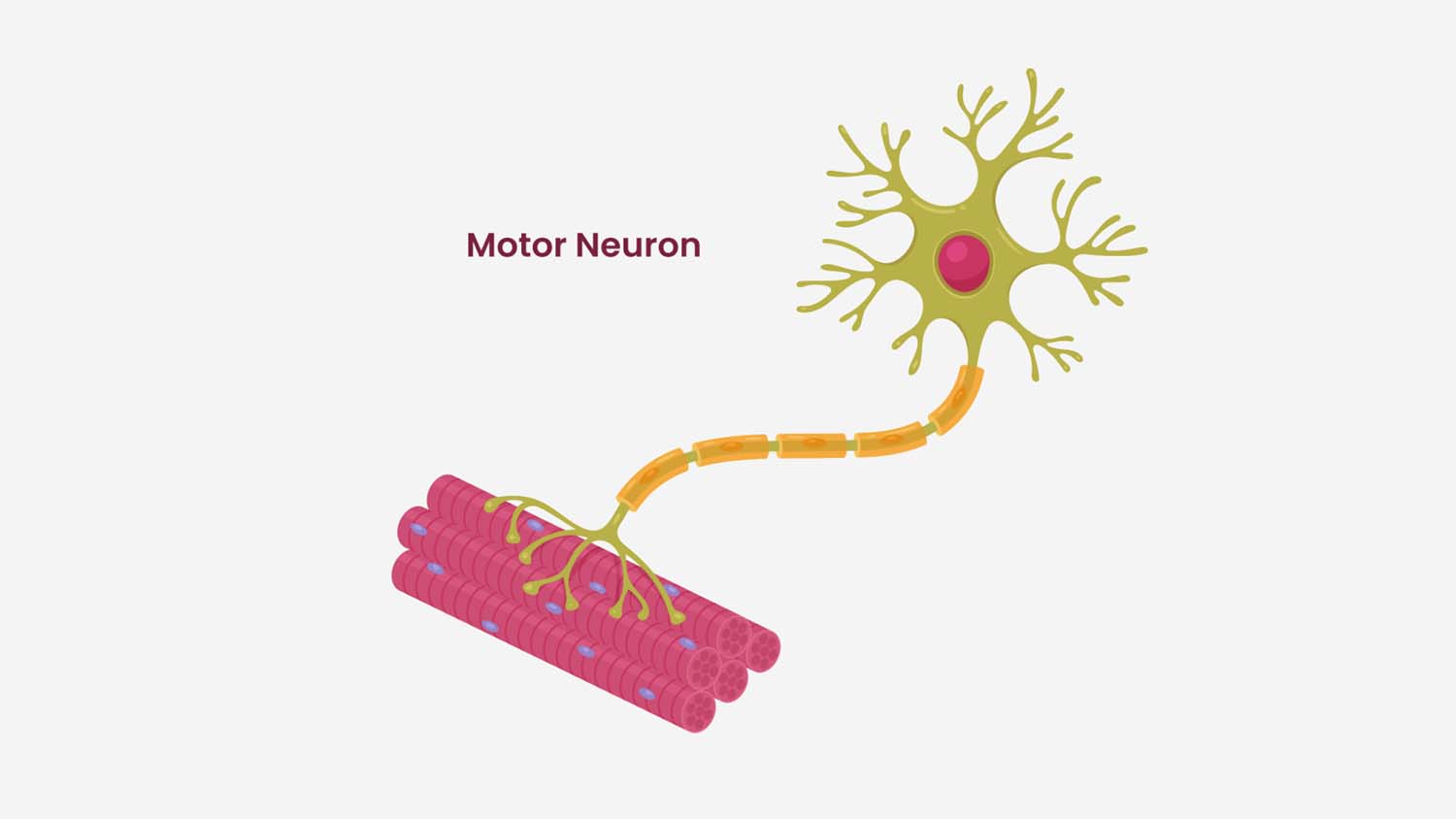
Acetylcholine Release at the Neuromuscular Junction
When the action potential reaches the synaptic bulb at the end of the axon, it activates voltage-gated calcium channels at around +30 millivolts. Calcium flows into the terminal.
Calcium binds to a protein called synaptotagmin, which acts as the calcium sensor on vesicles containing acetylcholine (ACh). This binding initiates a trans-SNARE complex, a four-helix protein structure formed by synaptobrevin (on the vesicle), syntaxin, and SNAP25 (on the target membrane). This complex drives vesicle fusion and exocytosis, releasing acetylcholine into the synaptic cleft.
Once the action potential ends and calcium stops entering, ACh is rapidly broken down by acetylcholinesterase into choline and acetate. Choline is taken back up into the synaptic bulb, where choline acetyltransferase combines it with another acetate to regenerate acetylcholine, which is then packaged back into vesicles via a proton transporter.
The Neuromuscular Junction: Triggering the Muscle
Acetylcholine crosses the synaptic cleft and binds to nicotinic acetylcholine receptors on the muscle cell membrane. These are ligand-gated, non-selective cation channels. They allow both sodium in and potassium out, but sodium influx dominates, depolarizing the muscle membrane and initiating an action potential that propagates along the muscle fiber.
This depolarization travels down into the T-tubules, deep invaginations of the muscle membrane that carry the signal into the interior of the cell. There it activates the dihydropyridine receptor (DHPR), an L-type calcium channel that, in skeletal muscle, acts as a mechanical sensor rather than an actual calcium channel. It is physically coupled to the ryanodine receptor (RYR1) on the sarcoplasmic reticulum. When the DHPR senses voltage, it mechanically opens the ryanodine receptor, releasing calcium into the sarcoplasm.
In cardiac muscle, this process differs. Calcium entering through the DHPR triggers calcium-induced calcium release through the ryanodine receptor, rather than direct mechanical coupling.
Cross-Bridge Cycling: From Calcium to Contraction
Released calcium binds to troponin C on the thin filament. The thin filament is composed of actin, with tropomyosin wrapped around it and the troponin complex (troponin I, C, and T) sitting on top. At rest, tropomyosin physically blocks the myosin binding sites on actin, preventing cross-bridge formation.
When calcium binds troponin C, it causes a conformational change. Troponin I, the inhibitory subunit bound to actin, moves aside, and tropomyosin shifts out of the groove, exposing the myosin binding sites.
Cross-bridge cycling then proceeds in this sequence:
- Rigor state: myosin is tightly bound to actin, with ADP already released. No movement yet.
- ATP binding: ATP binds to the myosin head, causing it to release from actin.
- Hydrolysis: the myosin ATPase hydrolyzes ATP to ADP + inorganic phosphate (Pi). This cocks the myosin head into a high-energy position. The head re-binds actin weakly.
- Power stroke: inorganic phosphate is released, triggering a conformational change that causes the myosin head to bind actin tightly and swing, pulling the actin filament (and the Z-disc it’s attached to) toward the M-line. The sarcomere shortens.
- ADP release: ADP leaves, returning the myosin to the rigor state. The cycle repeats as long as calcium and ATP are available.
It’s important to note: it’s not the hydrolysis of ATP that causes the power stroke. The hydrolysis cocks the head. The power stroke is driven by the release of inorganic phosphate and the subsequent conformational change.
The term rigor mortis comes directly from this cycle. At death, ATP is no longer available to release myosin from actin, so the cross-bridges remain locked in place.
Muscle Relaxation: Removing the Calcium
Contraction continues as long as calcium is present. To relax the muscle, calcium must be removed from the sarcoplasm.
SERCA pumps (sarcoplasmic/endoplasmic reticulum calcium ATPase pumps) continuously pump calcium back into the sarcoplasmic reticulum in exchange for hydrogen ions. This process uses ATP, and approximately 20 to 30 percent of the cell’s ATP budget goes to SERCA pumps alone. Inside the SR, the protein calsequestrin binds and sequesters over 90% of the stored calcium, keeping free calcium concentration low and maintaining the driving force for future release.
In type 2 (fast-twitch) muscle fibers, the protein parvalbumin plays an additional role. It rapidly binds free calcium in the sarcoplasm directly, accelerating the drop in calcium concentration and enabling faster relaxation, which is essential for generating rapid, repeated force.
The muscle membrane repolarizes via potassium efflux. Because the T-tubules are narrow, potassium can accumulate there and potentially impair repolarization, which is where chloride influx becomes important as a buffering mechanism.
Putting It All Together
The full sequence, from brain signal to muscle contraction to relaxation, involves:
Resting membrane potential established by the Na⁺/K⁺ ATPase, EPSPs and IPSPs integrated at the axon hillock, threshold reached at −55 mV, action potential fires, sodium channels open then inactivate, potassium channels repolarize and hyperpolarize the cell, saltatory conduction down the myelinated axon, calcium enters the synaptic bulb, synaptotagmin initiates SNARE complex, acetylcholine exocytosis, ACh binds nicotinic receptor, depolarization wave along muscle membrane, T-tubule voltage sensed by DHPR, ryanodine receptor opens, calcium floods sarcoplasm, calcium binds troponin C, tropomyosin shifts, myosin binds actin, power stroke, sarcomere shortens, SERCA pumps restore calcium, muscle relaxes.
Each step depends on the one before it. And every step has an energy cost, which is why understanding bioenergetics and membrane physiology aren’t separate topics. They’re the same story.
Tyler W. LeBaron, MSc., PhD.
Tyler W. LeBaron, MSc, PhD is a is a researcher and educator who translates complex science into practical insight on health, performance, and human potential. He is the Founder and Executive Director of the Molecular Hydrogen Institute (a science-based 501(c)3 nonprofit) and an adjunct professor of exercise physiology and chemistry at Southern Utah University. Tyler is known for evidence-based, engaging presentations that challenge assumptions, clarify emerging science, and inspire high-performing individuals and organizations. He has 80+ peer-reviewed publications, 2,000+ citations, top 0.5% recognition in oxidative stress, and has delivered invited talks on six continents.
Disclaimer: This blog is for general informational purposes only and does not constitute the practice of medicine, nursing or other professional health care services, including the giving of medical advice, and no doctor/patient relationship is formed. The use of information on this blog or materials linked from this blog is at the user’s own risk. The content of this blog is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Users should not disregard, or delay in obtaining, medical advice for any medical condition they may have, and should seek the assistance of their health care professionals for any such conditions.
You may also like these…